At the heart of Molecular Workbench’s modeling of atomic interactions is a profoundly important but fundamentally simple concept:
At close distances, atoms attract each other until they get so close that they repel.
Here’s a demo of that concept: two atoms interacting. Drag the green atom to various locations near and far from the purple atom and watch what happens as the two atoms approach each other and move apart. (If you’re wondering why the atom slows down and stops, the answer is that we apply an artificial damping force to the green atom in order to make it easier for you to “grab” it and play with it.)
This concept is called intermolecular attraction. Molecular Workbench (MW) uses an approximate formula for calculating the intermolecular potential that was originally proposed by John Lennard-Jones (in 1924!) and is now called the “Lennard-Jones potential” or L-J for short.
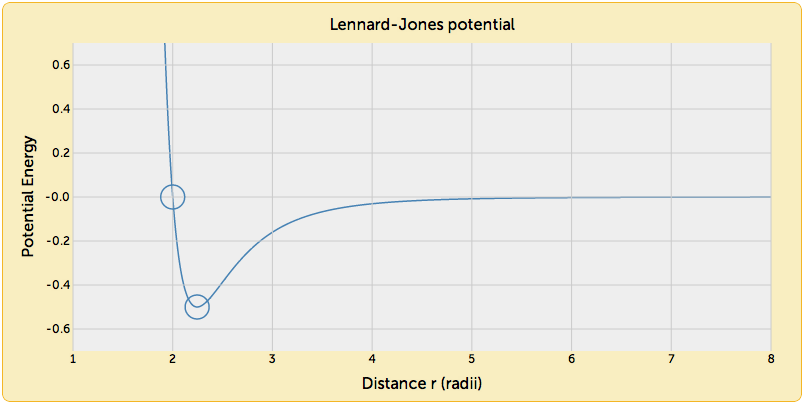
Here you see the L-J potential as a graph. The horizontal axis shows distance between two atoms, the vertical axis is the net intermolecular energy, with regions of negative slope indicating that the resulting force is repulsive, and regions of positive slope indicating attraction. This graph shows that these atoms will attract to each other if they are more than 2.3 radii apart, but begin to repel sharply at distances less than that value as shown by the steep rise in the curve.
This interaction is not just a fundamental concept in physics and chemistry. It also is quite central to the MW simulation engine. We typically do this calculation tens or hundreds of thousands times per second, especially when we have many atoms interacting, such as here:
If you study our code, going deeper and deeper, you’ll peel away layers of the coding onion, until you get to the very center of this model, which calculates the L-J force between just one pair of atoms. (Did we mention that the reason the L-J approximation is used is that it’s considered relatively fast to calculate?) It does this for each of the many pairs of interacting atoms, repeating over and over, with each time-tick of our simulation.
As it turns out, the L-J formula is still computationally demanding, requiring calculating 6th and 12th powers. (That’s the theory. In practice, the form that is most convenient for our code happens to use the 8th and 14th powers. That also speeds things up–but we’ll save that for another blog post.) So when we’re looking for ways to make our code run faster and our model run better, the L-J calculation is a prime place to look!
In our first pass at improving speed (in MW Classic), we converted the 6th and 12th power calculations to simpler repetitive multiplications:
X2 = X * X
X3 = X2 * X
X6 = X3 * X3
X12 = X6 * X6
That gave us just 4 multiplication tasks, instead of 16 (X6 has 5 multiplications, X12 has 11). This reduced the calculation demand to 25%. The model ran faster and smoother.
Recently, we tried another method to improve speed: using a look-up table, with pre-calculated values. We computed the L-J values for each element type for dozens of typical interatomic distances and put them into a table. We thought this would save computational time because the software could simply look up the values in the table without any multiplication.
We tested the two methods (multiplication vs. look-up table) over many iterations and found that the look-up table was slower! As we investigated further, we saw that accessing the look-up table had its own overhead in terms of use of cache memory and data transfer. This was evidence of a trend in improving computer speeds over the past few years: computation speeds improve at a faster rate than memory access speeds. They both are faster, but computation (i.e., all that multiplying we have to do) has gotten the improvement edge.
So, after this testing, we went back to the efficient multiplication approach. This illustrates our basic approach: creative thinking validated by empirical data.
We will continue to develop and test creative ways to speed the software and improve the user experience, especially as we move to support a greater variety of learning activities and computer platforms.