
Tim Erickson is an educator focusing on mathematics, physics, and data science; a consultant on several data science projects; and the author of Awash in Data. CODAP is great for exploring data. You can make graphs and do cool data moves, such as filtering, grouping, and summarizing your data. You can download data from public […]

The WATERS (Watershed Awareness using Technology and Environmental Research for Sustainability) project recently ended with a Master Teacher Workshop at our Concord office for selected teachers who participated in the National Science Foundation (NSF)-funded research with excellence and enthusiasm. The goal of the workshop was to exchange best practices for teaching the now freely available, […]
At the Concord Consortium, 12 of our research scientists are Principal Investigators or Co-Principal Investigators* on our dozens of National Science Foundation-funded projects. We polled our Principal investigators and their project managers to find out what it’s like to run a successful research and development project. Below are their top ten tips. 1. Like most […]

We are delighted to announce that Bridget Druken has been awarded our Robert F. Tinker Fellowship for 2024. Describing her background, Druken begins, “I loved all my classes and subjects in high school.” She then adds, “but really admired my math teachers.” She smiles as she runs down their list of names. She credits one […]
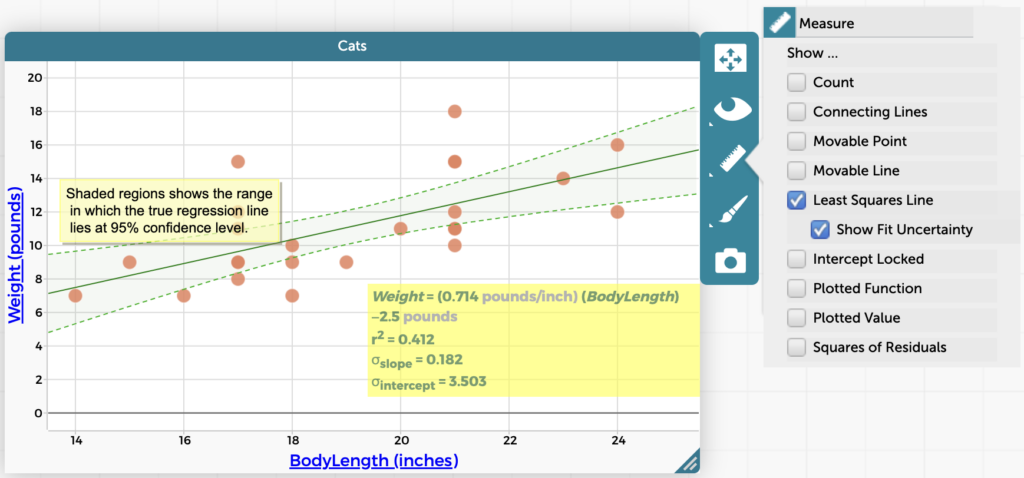
The CODAP website has a growing library of CODAP help resources aimed at highlighting handy features and improving your CODAP experience, so you can do even more with data exploration and visualization. Below you’ll find a sample of recent CODAP help resources and some additional CODAP features you may not have known about. Read on to […]
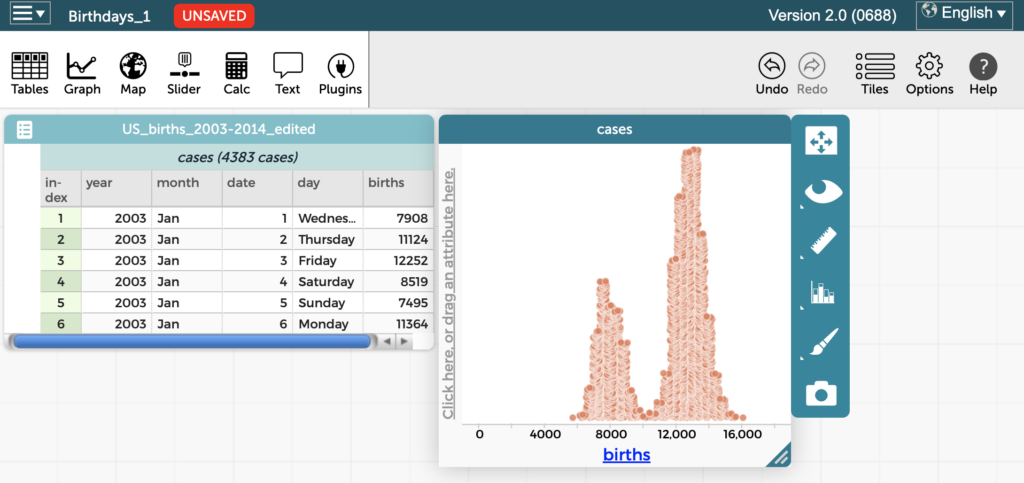
If one of your New Year’s resolutions is to add more data exploration to your lessons this year, we have a week’s worth of activities to get you started. Because data is interdisciplinary, you can find a home for it in any scientific discipline from astronomy to zoology, and across the halls in humanities, social […]

Last summer, thick plumes of wildfire smoke from northern Canada blanketed cities as far south as Washington, D.C., in an eerie orange haze. Record-breaking wildfires in provinces across Canada resulted in catastrophic damage to homes and communities, the destruction of more than 45.7 million acres of forest, and dangerously unhealthy air quality throughout the Northeast […]
The Concord Consortium, in collaboration with Texas Tech University, the University of Florida, and WestEd, was awarded a $4 million Education Innovation and Research grant from the U.S. Department of Education. The five-year project will develop a year-long Artificial Intelligence (AI) in Math supplemental certificate program for secondary Algebra I or Integrated Math 1 classes. […]

2024 marks our 30th anniversary. Our origins reach back to a simple beginning, on a single-board computer with a mere one kilobyte of memory. But it’s not about the computer itself—it’s never just the technology. It’s what the computer made possible that matters. When Bob Tinker connected a KIM-1 computer to an expansion board he’d […]
To work towards our mission to innovate and inspire equitable, large-scale improvements in STEM teaching and learning through technology, we make our STEM resources free and our research findings accessible and usable. Achieving such an ambitious mission takes countless partners and perspectives, and we are thrilled to collaborate with teachers, students, scientists, and researchers. In […]